Atomic orbital

In quantum mechanics and quantum chemistry, an atomic orbital is a function of an electron that describes the motion—in the quantum mechanical sense of the word—of the electron around the nucleus of an atom.
In the pre-computer era of quantum chemistry, an atomic orbital (AO) was seen as a solution of a one-electron Schrödinger eigenvalue equation for an atomic electron. In the case of one-electron atoms the Schrödinger equation is exactly solvable, that is, its solutions are known in analytic form. In the case of more-electron atoms the exact Schrödinger equation is not solvable. It must be replaced by an effective one-electron equation that contains the interaction between the electrons in an averaged manner; a single electron is considered that moves in a mean-field. A well-known mean-field method is the one proposed by Hartree and Fock. Because the effective equation depends only on the coordinates x, y, z of the single electron that is considered, the solution (eigenfunction) is a function of x, y, and z—an atomic orbital. If, in addition, the effective Schrödinger equation is invariant under rotation, like the Schrödinger equation of the one-electron atoms, it can be shown that the angular parts of the solutions consist of spherical harmonic functions. The latter functions determine the shape of the AOs.
Later in the development of quantum chemistry, especially in its computationally oriented branches, the term "atomic orbital" simply became to mean a one-electron function centered on an atom, not necessarily the solution of an effective one-electron atomic Schrödinger equation. For such functions it is meaningless to speak of their (orbital) energy. One-electron basis functions (briefly orbitals) are needed in many numerical methods for solving the Schrödinger equation of atoms and molecules. Functions of rA1 (the distance of electron 1 to nucleus A) are used as the radial parts of basis functions depending on rA1, the position vector of electron 1 with respect to a system of axes with origin on A. It is common to choose solid harmonic functions (which differ from spherical harmonics only by a factor rA1ℓ, the ℓth power of rA1) as the angular parts of the one-electron basis functions. Solid harmonics depend on the spherical polar coordinates of rA1.
Contents |
[edit] Solutions of the atomic Schrödinger equation
As stated in the introduction, the original meaning of atomic orbital is the quadratically integrable eigenfunction of a one-electron atomic Schrödinger equation. The eigenfunctions and eigenvalues of a one-electron Schrödinger equation are orbitals and orbital-energies, respectively. For a multi-electron atom, an independent-particle model leads to an effective one-electron Schrödinger equation, describing one electron in the mean field of all other electrons. That is, orbitals and orbital-energies are not only defined for physical one-electron systems, such as the H-atom, but also for N-electron systems by the aid of an independent-particle approximation.
Bohr[1] was the first to see how atomic orbitals can be used in an understanding of the Periodic table of elements. In Bohr's explanation of the Periodic table, the atomic orbitals of all elements carry the same labels as the exact hydrogen orbitals. That is, they have a principal quantum number n and a letter (s, p,...) designating the azimuthal quantum number (angular momentum quantum number) ℓ. Because Bohr's work predates the Schrödinger equation, Bohr did not know how the atomic orbitals could be computed, he simply postulated their existence and their quantum numbers.
Bohr's use of the quantum numbers n and ℓ as exact labels is not only legitimate for hydrogen-like atoms, but also for AOs that arise as solutions from a one-electron mean-field Schrödinger equation with a central (spherically symmetric) potential field. The central field approximation, which involves the angular averaging of the electronic mean field, is necessary to have ℓ as an exact quantum number.
Two major differences between an atomic effective one-electron model and the hydrogen-like (physical one-electron) atom are:
- The radial solutions are given numerically, and are no longer known analytic functions, such as the Laguerre functions for the hydrogen-like atom. The angular parts, however, are the same analytic functions (spherical harmonics) as in the case of the hydrogen-like-atom.
- The high degeneracy of the H-atom is lifted. In the hydrogen atom all orbitals of certain n are degenerate. For instance, the orbitals in the n = 4 shell: 4s, 4p, 4d, and 4f, all have the same energy (proportional to 1/n2 = 1/16). In the N-electron atom only the AOs of the same azimuthal quantum number ℓ are degenerate.
Note the role of the principal quantum number n in the effective one-electron model. It is simply an index that counts increasing orbital energies, starting at n = ℓ + 1 (to be in line with the hydrogen-like AOs). In summary, the atomic orbitals of any N-electron atom (N ≥ 1) are labeled by n (indicating the energy) and ℓ (indicating the angular momentum).
It is found[2] that the orbitals arising from the central field, independent-particle model applied to different atoms have the following order in increasing energy
- 1s, 2s, 2p, 3s, 3p, 3d, 4s, 4p, 4d, 5s, 5p, 6s, 4f, 5d, 6p, 7s, 5f, 6d.
For some atoms the order of 3d and 4s is flipped and for some other atoms the order of 4d and 5s is flipped. Note further that, for example 4f is not degenerate with 4d, which is the case for hydrogen-like atoms.
Knowing this order, one can, following Bohr, build up the atoms in the Periodic Table by the Aufbau (building-up) principle: fill orbitals in increasing energy. Doing this one must obey the Pauli exclusion principle that forbids more than two electrons per spatial orbital. Allowed is at most one electron with α spin and one with β spin. In addition recall that there are 2l+1 orbitals of certain l. For instance, the neon atom (atomic number Z = 10) has the electronic configuration (n = 1 and n = 2 shells are completely filled):
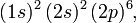
meaning two electrons are in the 1s, two electrons in the 2s, and six electrons in the 2p AOs. Similarly, chlorine (Z = 17) has
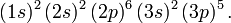
For more details the article Periodic table of elements may be consulted. A list of the electronic configurations of the ground states of the first 94 elements may be found here.
[edit] Formal definition of atomic-centered one-electron functions
An atomic orbital (in its modern definition as a nuclear-centered one-electron function) is a function depending on a single 3-dimensional vector rA1, which is a vector pointing from point A to electron 1. Generally there is a nucleus at A.[3] The following notations for an AO are frequently used,
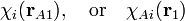
but other notations can be found in the literature. In the second notation the center A is added as an index to the orbital. We say that χA i (or, as the case may be, χi) is centered at A. In numerical computations AOs are either taken as Slater type orbitals (STOs) or Gaussian type orbitals (GTOs). Hydrogen-like orbitals are rarely applied in numerical calculations, because they are not complete and fairly difficult to handle.
The orbital is quadratically integrable (has finite norm), which means that the following integral is finite,
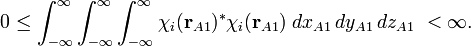
Its integrand being real and non-negative, the integral is real and non-negative. The integral is zero if and only if χ i is the zero function.
[edit] Atomic orbital basis sets
As stated above, in computational quantum chemistry atomic orbitals (in their meaning of atom-centered one-electron functions, not as solutions of an atomic Schrödinger equation) are used as mathematical building blocks to obtain approximations of N-electron atomic and molecular wave functions—solutions of the time-independent electronic Schrödinger equation.
A computation of a molecular wave function usually goes through the following steps:
- A geometry of the molecule is chosen (in accordance with the Born-Oppenheimer approximation the nuclei are clamped in space).
- A set of basis functions is chosen that are centered on the nuclei. Sometimes this AO set is augmented with orbitals in the middle of bonds, where there are no nuclei. These "bond-centered" basis functions are obviously not atomic orbitals. Preferably the basis set is as close as possible to a complete basis of one-electron Hilbert space,
![{\scriptstyle L^2[\mathbb{R}^3]}](../w/images/math/2/a/b/2ab6ccc126c561070a79b96ccddb1dfe.png) , but computer time is a practical limit. (Many methods require computer times proportional to n6 or n7, where n is the number of basis functions.)
, but computer time is a practical limit. (Many methods require computer times proportional to n6 or n7, where n is the number of basis functions.)
- An LCAO Hartree-Fock calculation yields the MO coefficients cA i and the same number of MOs as basis functions (namely n).
- The MOs are used in a post-Hartree-Fock calculation (configuration interaction, Møller-Plesset perturbation theory, coupled cluster theory, etc.).
The AOs and MOs spanning the very same orbital subspace of one-electron Hilbert space, it would be conceivable to skip the Hartree-Fock calculation. However, it turns out that the post-Hartree-Fock methods converge much better when they are based on MOs instead of on (orthogonalized) AOs.
The size of the basis is of crucial importance. The qualitative, pre-computer, MO-theoretical studies were invariably based on minimum basis sets. That is, only orbitals occupied in the free atoms were included in the basis. (But note that, for instance for the ground state boron atom with its electron configuration 1s22s22p, it cannot be said whether 2px, 2py, or 2pz is occupied. In such a case all three degenerate p orbitals are included in the minimum basis set). It was natural that the first computer calculations followed this pattern and applied minimum basis sets. However, it soon was found that such basis sets give very disappointing results. After this became clear in the late 1960s and early 1970s, search for good basis sets became an important subject of research.
For the mathematical definition of basis sets, we refer to the article Gauss type orbitals (GTOs), because the majority of present-day computer programs handle only GTOs. In the GTO article the concepts of primitive orbital and contracted set are introduced. In a minimum GTO basis set (also known as a single-zeta basis set) every atomic orbital occupied in the free atom is represented by a single contracted set. The term "single-zeta" is historic and refers back to the days that Slater type orbitals were universally used and to the fact that the screening constant in an STO is conventionally indicated by the Greek letter zeta (ζ). Single-zeta (SZ) basis sets giving poor quantitative results, the next step is the use of double-zeta (DZ) basis set, which involves a doubling of the SZ basis. Triple-zeta (TZ) (tripling of the minimum basis), quadruple-zeta (QZ), quintuple-zeta (5Z), sextuple-zeta (6Z) basis sets all have been proposed and have been constructed.
For instance a QZ GTO basis for HCN includes: 4 s-orbitals (s-type contracted sets) on H, 8 s-orbitals on both C and N, 4 px-, 4 py-, and 4 pz-orbitals on C and N. In all these cases the construction of the contracted set (a single one-electron basis function) involves the determination of the exponents of the primitive Gaussians and the corresponding contraction coefficients.
It is known that an atom polarizes (gets a dipole) under influence of an external electric field. It is also known that, in the presence of such a field, AOs of higher ℓ quantum numbers must be included in the basis in order to obtain reliable results. When, for instance, the ground state boron atom (1s22s22p) is polarized, there will be a mixing, linear in the strength of the external field, between 2p and 3d. That is, to describe the polarization of the boron atom correctly, one or more 3d sets (sets of five degenerate AOs) must be added to the basis of boron. Since in molecules strong electric fields, due to the nuclei, are present, it stands to reason that polarization functions improve the computational results. For an atom that only has occupied s orbitals, like hydrogen and helium, polarization functions start at ℓ = 1. For atoms such as boron with occupied p-orbitals, the polarization functions are by definition d, f, g, etc. orbitals.
Finally, it is emphasized that in the present-day elaborate computer calculations, an atomic orbital does not have the physical importance that was attached to it in pre-computer times. The basis functions are now looked upon as building bricks in a numerical approximation, no chemical or physical conclusions are to be drawn from the functions themselves. The relevant conclusions about nature emerge from the final computational results. Especially in the post Hartree-Fock methods, the contribution of the individual basis functions to the conclusions is deeply hidden in the output and not at all transparent.
[edit] Notes and references
- ↑ N. Bohr, Zeitschrift für Physik, vol. 9, p. 1 (1922)
- ↑ J. C. Slater, Quantum Theory of Atomic Structure, vol. I, McGraw-Hill, New York (1960), p. 193
- ↑ Floating orbitals and bond-centered functions, both of which are with respect to a point A where there is no nucleus, are sometimes used.