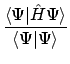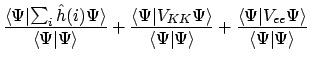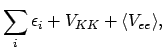Next: Dichtheidsmatrix
Up: Aantekeningen bij het college
Previous: Elektronen dichtheid
De effectieve operator in MO theorie, de Hartree-Fock methode
In MO-theorie proberen we de totale golffunctie te schrijven in termen van moleculaire orbitalen 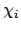 ,
die eigenfuncties zijn van een bepaalde effectieve één-elektron operator
,
die eigenfuncties zijn van een bepaalde effectieve één-elektron operator
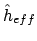 :
:
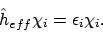 |
(18) |
De totale n-deeltjes golffunctie
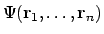 kunnen we dan schrijven in de vorm van een Slater-determinant als
kunnen we dan schrijven in de vorm van een Slater-determinant als
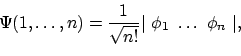 |
(19) |
waarin de 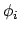 spinorbitalen (direct produkt van een ruimte-functie en een spinfunctie) zijn:
spinorbitalen (direct produkt van een ruimte-functie en een spinfunctie) zijn:
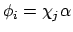 of
of
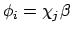 .
De verwachtingswaarde van de totale energie E berekenen we dan met behulp van de totale Hamiltoniaan:
.
De verwachtingswaarde van de totale energie E berekenen we dan met behulp van de totale Hamiltoniaan:
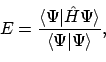 |
(20) |
en aangezien we nog weten dat deze verwachtingswaarde altijd hoger is dan de grondtoestandsenergie, is het dus de kunst om een
te vinden, zodanig dat de resulterende E zo laag mogelijk is.
Stel nu dat 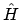 te schrijven was als een som van effectieve één-elektron operatoren:
te schrijven was als een som van effectieve één-elektron operatoren:
waarop we alwéér hoofdsstuk 3 los kunnen laten. De totale energie zou voor orthonormale spinorbitalen dan gegeven worden door
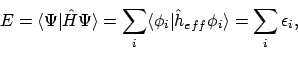 |
(22) |
ofwel, de totale energie zou simpelweg de som zijn van de orbitaal-energieën.
Helaas valt in de harde werkelijkheid de Hamiltoniaan niet te schrijven als een som van effectieve één-elektron operatoren. We weten immers dat deze (in de Born-Oppenheimer benadering) gegeven wordt door
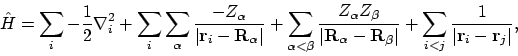 |
(23) |
waarin 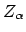 de lading van kern
de lading van kern 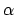 is, en
is, en
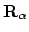 en
ri de postities van respectievelijk kern
en elektron i aanduiden. Aangezien we in de Born-Oppenheimer benadering de kernen hebben vastgezet is de kern-kern repuslie VKK, de derde term in de Hamiltoniaan, gewoon een vast getal. Verder hangt elke term in de eerste twee sommaties in de Hamiltoniaan van maar één elektron af; deze kunnen we dus samenpakken tot één-elektron Hamiltonianen
en
ri de postities van respectievelijk kern
en elektron i aanduiden. Aangezien we in de Born-Oppenheimer benadering de kernen hebben vastgezet is de kern-kern repuslie VKK, de derde term in de Hamiltoniaan, gewoon een vast getal. Verder hangt elke term in de eerste twee sommaties in de Hamiltoniaan van maar één elektron af; deze kunnen we dus samenpakken tot één-elektron Hamiltonianen
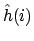 .
De laatste term, de elektron-elektron interactie Vee verstoort echter onze één-elektron aanpak, omdat elke term daar van twee elektronen tegelijkertijd afhangt. Schrijven we dus
.
De laatste term, de elektron-elektron interactie Vee verstoort echter onze één-elektron aanpak, omdat elke term daar van twee elektronen tegelijkertijd afhangt. Schrijven we dus
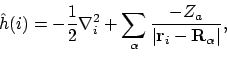 |
(24) |
dan wordt de Hamiltoniaan
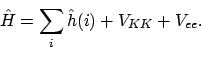 |
(25) |
De eerste term wordt ook wel de ``bare nucleus'' Hamiltoniaan genoemd, omdat hierin het effect van afscherming van de kern door andere elektronen niet meegenomen wordt. Als we die gebruiken als effectieve Hamiltoniaan om MO's te construeren:
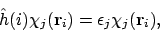 |
(26) |
dan wordt de verwachtingswaarde van de totale energie dus gegeven door
ofwel de sum van de orbitaal-energieën, de kern-kern repulsie, en een correctieterm
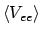 .
Hieraan zien we dat het Aufbau-principe, dat zegt dat de totale energie minimaal is als we de elektronen in de orbitalen met de laagste orbitaal-energie stoppen, klopt zolang de correctie als gevolg van de elektronen-interactie klein is.
.
Hieraan zien we dat het Aufbau-principe, dat zegt dat de totale energie minimaal is als we de elektronen in de orbitalen met de laagste orbitaal-energie stoppen, klopt zolang de correctie als gevolg van de elektronen-interactie klein is.
Het negeren van alle interactie met andere elektronen bij het construeren van de moleculaire orbitalen is natuurlijk niet erg fysisch. We willen daarom proberen een betere
te maken, door een effectieve potentiaal Veff bij de bare nucleus Hamiltoniaan op te tellen die deze interactie zo goed mogelijk beschrijft:
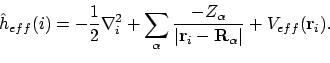 |
(28) |
De vraag is nu natuurlijk: hoe maken we Veff? Stel dat we de interactie van elektron i met elektron j willen beschrijven, en dat we de elektronendichtheidsverdeling
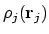 van j weten. De hoeveelheid lading ten gevolge van elektron j in een blokje
drj om de plaats
rj is dan
van j weten. De hoeveelheid lading ten gevolge van elektron j in een blokje
drj om de plaats
rj is dan
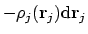 .
Stel elektron i zit op plaats
ri, dan kunnen we de totale Coulomb-interactie met elektron j dus uitrekenen door het optellen van de Coulomb interactie met elk van de blokjes apart, ofwel, voor oneindig kleine blokjes
.
Stel elektron i zit op plaats
ri, dan kunnen we de totale Coulomb-interactie met elektron j dus uitrekenen door het optellen van de Coulomb interactie met elk van de blokjes apart, ofwel, voor oneindig kleine blokjes
![\begin{displaymath}V_{eff,j}(\mathbf{r}_i)
= \int \frac{[-1][-\rho_j(\mathbf{r...
...\vert\mathbf{r}_i - \mathbf{r}_j\vert}\mathrm{d}\mathbf{r}_j .
\end{displaymath}](img94.gif) |
(29) |
De totale Veff krijgen we dan door de interactie met alle elektronen mee te nemen:
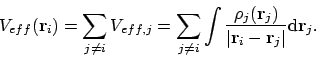 |
(30) |
Het enige probleem zit nu nog in 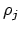 .
Deze is immers gedefinieerd als
.
Deze is immers gedefinieerd als
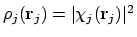 .
Dus om de elektronendichtheid van elektron j te weten, hebben we de orbitaal nodig waarin j zit. Maar deze orbitalen proberen we juist uit te rekenen met
.
We lossen dit op door middel van een iteratief proces: we maken een gok voor de MO's (bijvoorbeeld door eigenfuncties van de bare nucleus Hamiltoniaan te nemen), construeren hieruit een
,
berekenen hieruit de nieuwe MO's, vullen deze weer in om een nieuwe
uit te rekenen, etc. totdat de MO's niet meer veranderen.
.
Dus om de elektronendichtheid van elektron j te weten, hebben we de orbitaal nodig waarin j zit. Maar deze orbitalen proberen we juist uit te rekenen met
.
We lossen dit op door middel van een iteratief proces: we maken een gok voor de MO's (bijvoorbeeld door eigenfuncties van de bare nucleus Hamiltoniaan te nemen), construeren hieruit een
,
berekenen hieruit de nieuwe MO's, vullen deze weer in om een nieuwe
uit te rekenen, etc. totdat de MO's niet meer veranderen.
In bovenstaande redenering is geen rekening gehouden met de anti-symmetrie van de golffunctie ten opzichte van verwisseling van twee elektronen. Omdat we ieder elektron in één specifieke spinorbitaal gestopt hebben, heeft dit tot gevolg dat de effectieve één-elektron Hamiltoniaan voor ieder elektron anders is. Als we het goed zouden doen, zouden we dus een effectieve operator moeten construeren, uitgaande van een Slater-determinant golffunctie. Dit kan gedaan worden door de totale energie als functie van de MO's te minimaliseren. Aangezien dit tamelijk moeilijk is, zullen we hier alleen enkele opmerkingen over het resultaat maken. Als we de MO's schrijven als een lineaire combinatie van atomaire orbitalen 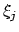 :
:
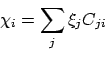 |
(31) |
dan worden de optimale expansiecoëfficiënten verkregen door de matrix vergelijking
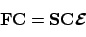 |
(32) |
op te lossen. Hierin is
F de Fock-matrix,
S de bekende overlap-matrix, en
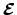 de diagonaalmatrix met orbitaal-energieën. De Fock-matrix is de matrix van de effectieve één-elektron Hamiltoniaan in een basis van atomaire orbitalen, en hangt af van de coëfficiënten
C. Voor iedere moleculaire orbitaal (kolom in de
C-matrix) hebben we dus
de diagonaalmatrix met orbitaal-energieën. De Fock-matrix is de matrix van de effectieve één-elektron Hamiltoniaan in een basis van atomaire orbitalen, en hangt af van de coëfficiënten
C. Voor iedere moleculaire orbitaal (kolom in de
C-matrix) hebben we dus
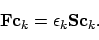 |
(33) |
Het enige verschil met een gewoon eigenwaardeprobleem is dat de Fock-matrix afhangt van de MO's. Oplossen van deze vergelijkingen gebeurt dus weer door te itereren todat de MO's geconvergeerd zijn: de Hartree-Fock methode.
Next: Dichtheidsmatrix
Up: Aantekeningen bij het college
Previous: Elektronen dichtheid
Gerrit Groenenboom
2005-06-01