
Molden has its own force field module Ambfor. This is an external
program that can be started from Molden. Ambfor supports the protein
force field Amber and the small molecule force field GAFF (General Amber Force Field). These force fields
can be mixed. If you have a protein molden automatically types the protein
atoms with the Amber force field. The small molecule can be typed with the
GAFF force field. Amber and GAFF use atomic charges. For Amber they are
statically encoded into the Ambfor module. For the small molecule
the charges have to supplied separately. Quantum Chemically derived charges
are recommended for the GAFF force field, but since these are computationally
very expensive, you can also use Semi-Empirical derived charges (Mopac) with a
correction scheme, AM1BCC. AM1BCC is part of the antechamber
suite of programs.
In our example below we will use computationally cheap
charges, directly available from Molden. the files used in this tutorial are
available from the molden source distribution in the test directory.
First we read in the protein file dock_prot.pdb.
This is not a full protein, but only the part around the active site.
This file was used to dock small molecules into with FlexX.
Second, we will read in the docking result file dock_lig.mol2.
This file contains several poses of a docked ligand. When reading in this
file a Multi-mol file window will popup.
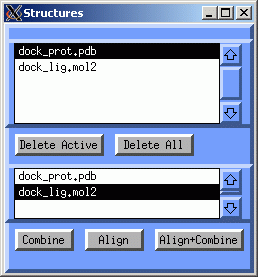
Now we will merge the protein with a docked ligand pose: In the Structures window, select dock_prot.pdb in the top listbox and select dock_lig.mol2 in the lower listbox and click Combine:
|
Zoom in to the drawing screen (mouse wheel up) and click with the second (or middle) mouse button on an atom of the ligand and select Calculate Charges.
Let Molden apply the atom types according to the GAFF force field. Click on
the force field icon  (top icon, middle column) in the molden control window.
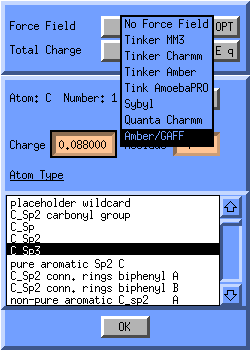
The Atom Attributes Window will pop up, click on the button next to
the text Force Field, and select Amber/GAFF:
(top icon, middle column) in the molden control window.
The Atom Attributes Window will pop up, click on the button next to
the text Force Field, and select Amber/GAFF:
|
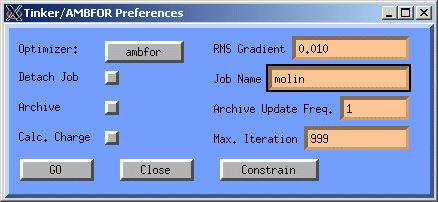
We are now ready to optimise the molecule: Click on the OPT button in the Atom Attributes Window. The Tinker/AMBFOR Preferences window will appear. Since we have already assigned charges, lets deactivate the automatic calculation of charges, the button next to the Calc. Charge text.
|
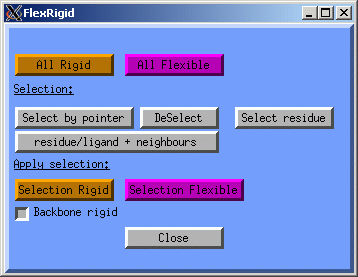
Since we do not have the full protein, we only want to optimise the part directly in contact with the ligand and the ligand itself, while keeping the rest rigid. Of the flexible residues we will keep the backbone atoms fixed. Click on the Constrain button, now the FlexRigid window will be displayed:
|
Now in the Tinker/AMBFOR Preferences window click on GO to start the partial optimisation.