
Molden has its own force field module Ambfor. This is an external
program that can be started from Molden. Ambfor supports the protein
force field Amber and the small molecule force field GAFF (General Amber Force Field). These force fields
can be mixed. If you have a protein molden automatically types the protein
atoms with the Amber force field. The small molecule can be typed with the
GAFF force field. Amber and GAFF use atomic charges. For Amber they are
statically encoded into the Ambfor module. For the small molecule
the charges have to supplied separately. Quantum Chemically derived charges
are recommended for the GAFF force field, but since these are computationally
very expensive, you can also use Semi-Empirical derived charges (Mopac) with a
correction scheme, AM1BCC. AM1BCC is part of the antechamber
suite of programs.
In our example below we will use computationally cheap
charges, directly available from Molden.
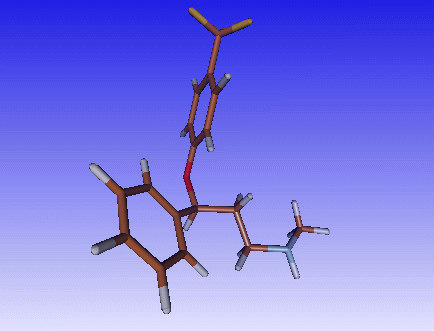
We will start loading a small molecule into Molden, I have chosen fluoxetine, an popular anti-depressant.
|
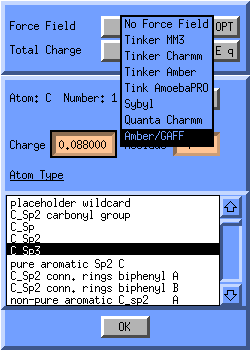
Let Molden apply the atom types according to the GAFF force field. Click on
the force field icon  (top icon, middle column) in the molden control window.
The Atom Attributes Window will pop up, click on the button next to
the text Force Field, and select Amber/GAFF:
(top icon, middle column) in the molden control window.
The Atom Attributes Window will pop up, click on the button next to
the text Force Field, and select Amber/GAFF:
|
You can see which atom types molden has assigned by activating Labels
--> ForceF.Type in the Molden control window.
If you are not happy with the assigned atom type or if the atom type is
missing, click on the atom in the molecular display and select a new
atom type from the list in the Atom Attributes Window.
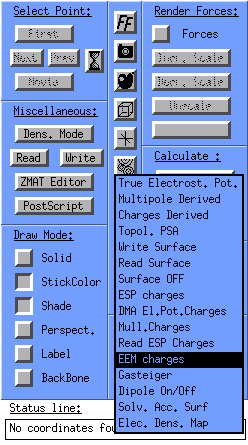
Let us calculate some charges for this molecule: Click on the surfaces/charges
 icon in the Molden control window and select EEM charges -> EEM Mull.
icon in the Molden control window and select EEM charges -> EEM Mull.
|
You can see what charges molden has assigned by activating Labels --> atom+charge in the Molden control window.
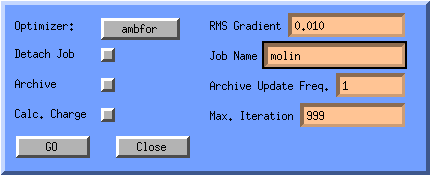
We are now ready to optimise the molecule: Click on the OPT button in the Atom Attributes Window. The Tinker/AMBFOR Preferences window will appear. Since we have already assigned charges, lets deactivate the automatic calculation of charges, the button next to the Calc. Charge text.
|
You can specify a gradient at which the molecule is considered optimised.
The lower the gradient specified, the more iterations Ambfor will need to
optimise the molecule. You can also specify the maximum iterations ambfor
can spend optimizing this molecule (Max. Iteration).
You can also Archive the optimisation. Ambfor will output the
intermediate structures every few iterations, into a file with the name
Job Name.arc. You can read this file into molden and replay
the optimisation by clicking the movie button in the molden control window.
To start the optimisation, click on the GO button. You can follow this optimisation in the molecular display. It can be aborted by hitting the escape key. This can also be used to stop the updating of the display when the ambfor program has already finished optimising the molecule. The updating of the display is usually running behind with the actual optimisation.
When the optimisation is finished, a window will pop-up:

|
Click OK and look at the optimized structure:
|
Problems you can run in to: