dutch
In opdracht 4 waren de matrix elementen van een effectieve één-elektron Hamiltoniaan voor B2 gegeven. De expliciete vorm van deze effectieve één-elektron Hamiltoniaan, de zogeheten Hartree-Fock-operator is gegeven in Engel, paragrafen 10.5 en 16.3. In deze opdracht gaan we de Fock operator construeren voor het B2molecuul en iteratief de (zelf-consistente) orbital energieën berekenen in het MO model. We beginnen met de ``core hamiltonian''. Voor de AO basis kiezen we een zogenaamde sto-3g basis. In deze basis wordt iedere AO gerepresenteerd als een (vaste) lineaire combinatie van drie Gaussische orbitalen (zie ook opdracht 2). We maken gebruik van een aantal gegeven MATLAB routines voor berekeningen met Gaussische basis sets.
B1=1 B2=2We kunnen een basis voor B2 als volgt opgeven:
name='sto3g'
basis=[ bas_lib('B',B1, 's', name)
bas_lib('B',B1,'px', name)
bas_lib('B',B1,'py', name)
bas_lib('B',B1,'pz', name)
bas_lib('B',B2, 's', name)
bas_lib('B',B2,'px', name)
bas_lib('B',B2,'py', name)
bas_lib('B',B2,'pz', name) ];
Dit geeft een basis bestaande uit 10 AOs omdat er in een
sto-3g basis voor het B atoom twee s-orbitalen gedefinieerd zijn.
De informatie wordt opgeslagen in een zogenaamd ``structure array''.
Als je bijvoorbeeld intypt basis(8) krijg je de details
van basis functie 8 te zien:
>> basis(8)
ans=
ipos: 2
itype: [1 0 0]
alpha: [2.2370 0.5198 0.1691]
c: [0.6080 0.3823 0.0606]
c0: [0.1559 0.6077 0.3920]
ng: 3
Dit definieert een zogenaamde gecontraheerde Gaussian van de vorm:
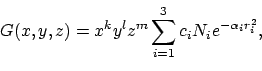 |
(1) |
basis(8).ipos=2 geeft aan dat x,y en z gedefineerd zijn
ten opzichte van kern 2, de machten k,l en m zijn gegeven
in het veld basis(8).itype ([1 0 0] is dus een px orbitaal).
De exponenten basis(8).alpha en de contractie coefficienten
basis(8).c0 kun je voor een heleboel Gaussian basis sets en atomen
vinden in een online database http://wserv1.dl.ac.uk:800/emsl_pnl/basisform.html
Het veld basis(8).c bevat de producten ci Ni.
De kernladingen definieren we met:
clear Z % voor de zekerheid Z(B1)=5 Z(B2)=5en de posities van de atoomkernen met:
R=3
Xatom=[ 0 0
0 0
-R/2 R/2]
Alle benodigde één-elektron integralen kunnen nu worden
berekend met
H1 = do_onelec(basis,Xatom,Z)Het structure
H1 bevat allerlei tussenresultaten die
handig zijn in verdere berekeningen. Voor het opstellen van
de core hamiltonian matrix hebben we alleen de
één-elektron kinetische energiematrix H1.T en
en de elektron-kern attractie matrix elementen H1.K nodig:
H0 = H1.T+H1.KDe overlap matrix zit in
H1.S. De MOs en orbital energiën
behorende bij de core hamiltonian zijn dus te vinden met:
S = H1.S; [C0,e0] = g_diag(H0,S);
C0 en e0.
Wat is er allemaal vreemd aan deze ``effectieve'' Hamiltoniaan?
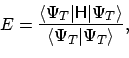 |
(2) |
do_twoelec en
energy_F nodig. De eerste routine rekent alle benodigde integralen over
de Coulomb operator
H1 nodig, waar alle informatie over de AO basis in zit:
ERI= do_twoelec(H1);Dit kost een paar minuten rekentijd, dus zorg ervoor dat je dit niet te vaak opnieuw doet. De energie kun je nu berekenen met:
[E,F]=energy_F(H1,C0,ndubbel,nenkel,ERI);waarbij
C0 de 10x10 matrix is waarvan iedere kolom bij een MO hoort.
Om een triplet-determinant te maken zet je ndubbel=4 en
nenkel=2, d.w.z., de MOs C0(:,[1 2 3 4]) krijgen ieder twee
elektronen en de MOs C0(:,[5 6]) krijgen ieder één elektron.
De energie komt terug in E. Let op: je moet er zelf nog de
kern-kern repulsie bij optellen.
Behalve de energie geeft de routine energy_F ook een verbeterde
effectieve één-elektron-matrix terug, de volledige Fock-matrix
F.
Dit is de matrix van de Fock operator die behalve de core hamiltonian
ook de Coulomb en exchange operatoren bevat, zie Engel, paragrafen
10.5 en 16.3. In deze Fock-matrix is rekening gehouden met de gegeven
orbital bezetting.
F, i.p.v. de core Hamilton matrix H0,
en maak een nieuw MO schema.C0 en stop de
nieuwe orbitalen in de variabele C:
[C,e] = g_diag(F, S);
[E,F]=energy_F(H1,C,4,2,ERI);
energie=E
for i=2:10
[C,e] = g_diag(F, S);
[E,F] = energy_F(H1,C,4,2,ERI);
energie(i,1)=E
end
C0. Je kunt een andere toestand krijgen door
de MOs zelf in een andere volgorde te zetten (dus niet volgens het
Aufbau principe), b.v.:
C=C0(:,[1:3 5 6 4 7:10])