In opdracht I waren de matrix elementen van een effectieve Hamiltoniaan voor B2 gegeven. In deze opdracht gaan we zelf een effectieve Hamiltoniaan construeren. We beginnen met de ``bare nucleus hamiltonian''. Voor de AO basis kiezen we een zogenaamde sto-3g basis. In deze basis wordt iedere AO gerepresenteerd als een (vaste) lineaire combinatie van drie Gaussische orbitalen (zie ook opdracht 4 van CB I). We maken gebruik van een aantal gegeven MATLAB routines voor berekeningen met Gaussische basis sets.
B1=1 B2=2We kunnen een basis voor B2 als volgt opgeven:
name='sto3g'
basis=[ bas_lib('B',B1, 's', name)
bas_lib('B',B1,'px', name)
bas_lib('B',B1,'py', name)
bas_lib('B',B1,'pz', name)
bas_lib('B',B2, 's', name)
bas_lib('B',B2,'px', name)
bas_lib('B',B2,'py', name)
bas_lib('B',B2,'pz', name) ];
Dit geeft een basis bestaande uit 10 AOs omdat er in een
sto-3g basis voor het B atoom twee s-orbitalen gedefinieerd zijn.
De informatie wordt opgeslagen in een zogenaamd ``structure array''.
Als je bijvoorbeeld intypt basis(8) krijg je de details
van basis functie 8 te zien:
>> basis(8)
ans=
ipos: 2
itype: [1 0 0]
alpha: [2.2370 0.5198 0.1691]
c: [0.6080 0.3823 0.0606]
c0: [0.1559 0.6077 0.3920]
ng: 3
Dit definieert een zogenaamde contracted Gaussian van de vorm:
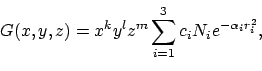 |
(1) |
basis(8).ipos=2 geeft aan dat x,y en z gedefineerd zijn
ten opzichte van kern 2, de machten k,l en m zijn gegeven
in het veld basis(8).itype ([1 0 0] is dus een px orbitaal).
De exponenten basis(8).alpha en de contractie coefficienten
basis(8).c0 kun je voor een heleboel Gaussian basis sets en atomen
vinden in een online database http://wserv1.dl.ac.uk:800/emsl_pnl/basisform.html
Het veld basis(8).c bevat de producten ci Ni.
De kernladingen definieren we met:
clear Z % voor de zekerheid Z(B1)=5 Z(B2)=5en de posities van de atoomkernen met:
R=3
Xatom=[ 0 0
0 0
-R/2 R/2]
Alle benodigde één elektron integralen kunnen nu worden
berekend met
H1 = do_onelec(basis,Xatom,Z)Het structure
H1 bevat allerlei tussenresultaten die
handig zijn in verdere berekeningen. Voor het opstellen van
de bare-nucleus hamiltonian matrix hebben we alleen de
één elektron kinetische energiematrix H1.T en
en de elektron-kern attractie matrix elementen H1.K nodig:
H0 = H1.T+H1.KDe overlap matrix zit in
H1.S. De MOs en orbital energiën
behorende bij de bare-nucleus hamiltonian zijn dus te vinden met:
S = H1.S; [C0,e0] = g_diag(H0,S);
C0 en e0.
Wat is er allemaal vreemd aan deze ``effectieve'' Hamiltoniaan?
 |
(2) |
do_twoelec en
energy_F nodig. De eerste routine rekent alle benodigde integralen over
de Coulomb operator
H1 nodig, waar alle informatie over de AO basis in zit:
ERI= do_twoelec(H1);Dit kost een paar minuten rekentijd, dus zorg ervoor dat je dit niet te vaak opnieuw doet. De energie kun je nu berekenen met:
[E,F]=energy_F(H1,C0,ndubbel,nenkel,ERI);waarbij
C0 de 10x10 matrix is waarvan iedere kolom bij een MO hoort.
Om een triplet-determinant te maken zet je ndubbel=4 en
nenkel=2, d.w.z., de MOs C0(:,[1 2 3 4]) krijgen ieder twee
elektronen en de MOs C0(:,[5 6]) krijgen ieder één elektron.
De energie komt terug in E. Let op: je moet er zelf nog de
kern-kern repulsie bij optellen.
Behalve de energie geeft de routine energy_F ook een verbeterde
effectieve één elektron-matrix (de Fock-matrix) F terug.
In deze Fock-matrix is rekening gehouden met de gegeven orbital
bezetting.
F, i.p.v. de bare-nucleus Hamilton matrix H0,
en maak een nieuw MO schema.C0 en stop de
nieuwe orbitalen in de variabele C:
[C,e] = g_diag(F, S);
[E,F]=energy_F(H1,C,4,2,ERI);
energie=E
for i=2:10
[C,e] = g_diag(F, S);
[E,F] = energy_F(H1,C,4,2,ERI);
energie(i,1)=E
end
C0. Je kunt een andere toestand krijgen door
de MOs zelf in een andere volgorde te zetten (dus niet volgens het
Aufbau principe), b.v.:
C=C0(:,[1:3 5 6 4 7:10])