Research
My research interest is in the computational modeling of the dynamics and physical properties of molecular systems in the solid state. Within this context, I especially like to focus on the algorithms, the implementation, and the development of the modeling techniques and the description of the interactions in the system.
Research projects
Some specific research projects in which I was involved were:- Unbiased, off-lattice kinetic Monte Carlo modeling of rare event dynamics in molecular ices,
- Surface diffusion of CO and CO2,
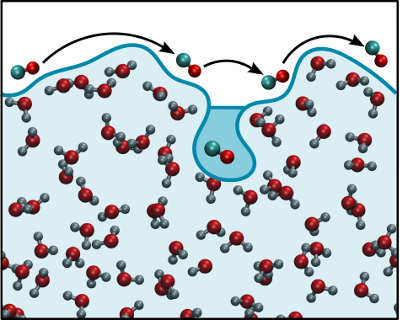
- Wetting behavior and many body interactions of adsorbed CO2 on water ice,
- Modeling of isothermal desorption experiments,
- Formation and evolution of structural defects on the surface of ice 1h,
- Lattice dynamics of graphite and graphene,
- Metropolis Monte Carlo simulations of the thermal expansion of carbon allotropes.
Ph.D. Research
 The subject of my Ph.D. project is the computational modeling of long-timescale dynamics in molecular ices at low temperatures. This research is motivated by the chemistry which is observed in interstellar molecular clouds. Here these ices play a crucial catalytic role in the formation of molecular species like hydrogen, water, and carbon dioxide, even before stars and planets have started to form. To understand these processes however, a good understanding of the structure and dynamics of the ice mantles is required.
The subject of my Ph.D. project is the computational modeling of long-timescale dynamics in molecular ices at low temperatures. This research is motivated by the chemistry which is observed in interstellar molecular clouds. Here these ices play a crucial catalytic role in the formation of molecular species like hydrogen, water, and carbon dioxide, even before stars and planets have started to form. To understand these processes however, a good understanding of the structure and dynamics of the ice mantles is required.
The challenge of this project is to simulate the system over very long timescales, required by the low temperatures in space, whilst keeping an atomistically detailed view of the system. To achieve this goal, we make us of a combination of publicly available and self-developed force fields to describe the interactions between molecules. These force fields are designed to reproduce experimental results and quantum-chemical ab-initio calculations at a very high computational efficiency. A description of the model and a C++ implementation, for H2O, CO, and CO2 is available in the code section.
Through the use of this set of force fields, we are able to model the dynamical processes in molecular ices at temperatures as low as 15 K reaching timescales on the order of seconds, far beyond the reach of conventional molecular dynamics simulations.
Master Research
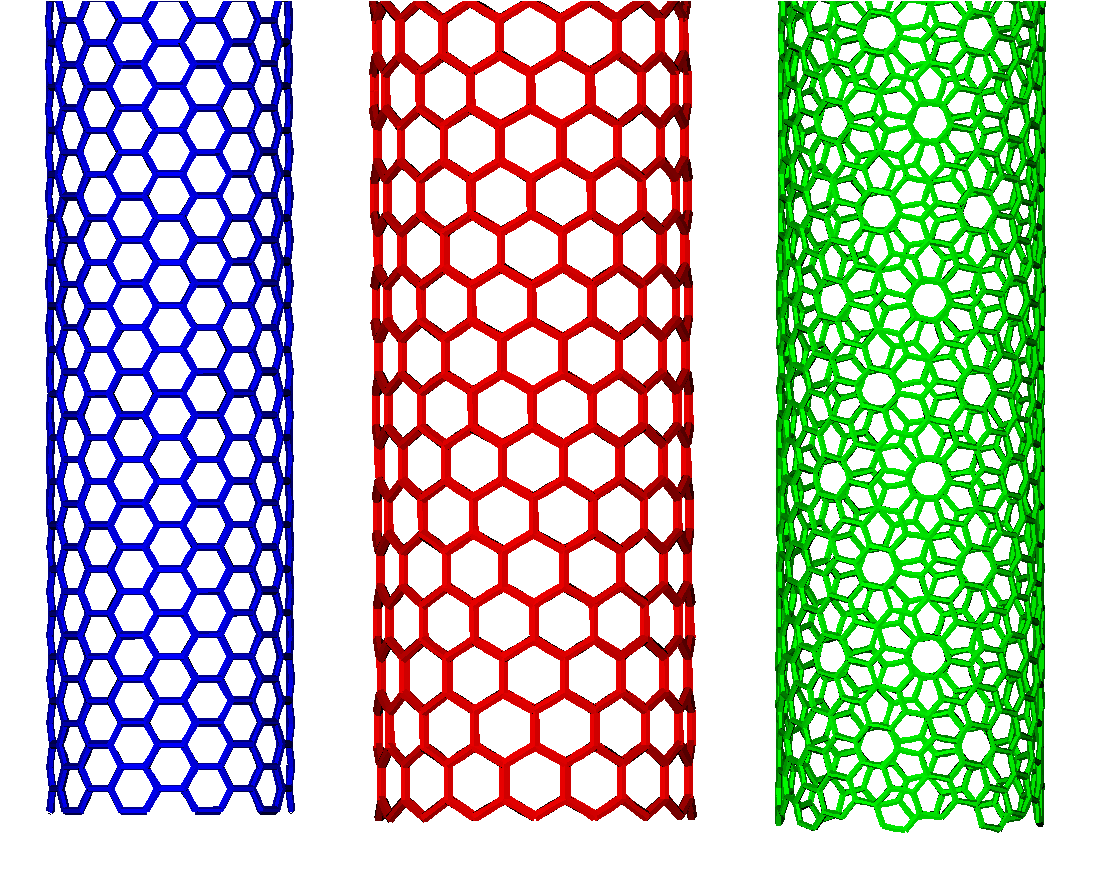
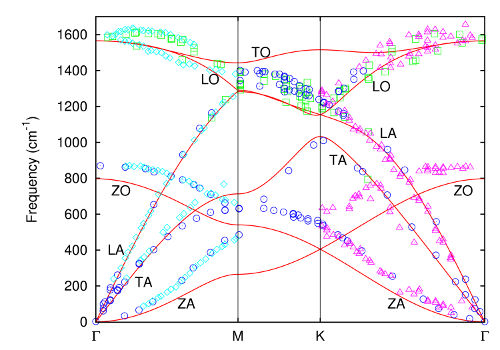 During my master internship at the theory of condensed matter group at the Radboud University, I performed Monte Carlo and lattice dynamical calculations of various carbon allotropes using an empirical bond-order potential, LCBOPII. My master thesis is available online.
During my master internship at the theory of condensed matter group at the Radboud University, I performed Monte Carlo and lattice dynamical calculations of various carbon allotropes using an empirical bond-order potential, LCBOPII. My master thesis is available online.