Next: 32.6 Options for selection
Up: 32 LOCAL CORRELATION TREATMENTS
Previous: 32.4 Summary of directives
- LOCAL=local
- Determines which method is used:
LOCAL=0: Conventional (non-local) calculation.
LOCAL=1: Local method is simulated using canonical
MOs. The local basis is used only at an intermediate stage
to update the amplitudes in each iteration (only for testing).
LOCAL=2: Calculation is done in local basis, but
without using local blocking (i.e. full matrices are used).
This is the most expensive method and only for testing.
LOCAL=3: Fully local calculation (obsolete).
LOCAL=4: Fully local calculation (default). This is the fastest
method for large molecules with many weak pairs and requires minimum memory.
- PIPEK=option
- If this option is given and option
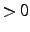 ,
the orbitals are localized using the Pipek-Mezey technique. If this option is
not given or option=0 (default), the orbitals are localized unless localized orbitals are found in
the orbital record (cf. ORBITAL directive and LOCALI command).
In the latter case, the most recent localized orbitals are used.
Setting option=-1 switches the localization off.
If option
,
the orbitals are localized using the Pipek-Mezey technique. If this option is
not given or option=0 (default), the orbitals are localized unless localized orbitals are found in
the orbital record (cf. ORBITAL directive and LOCALI command).
In the latter case, the most recent localized orbitals are used.
Setting option=-1 switches the localization off.
If option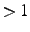 the localized orbitals are printed. Note: Boys localization
can only be performed using the LOCALI command. The program will use
the Boys orbitals if they are found in the orbital record and the PIPEK option
is absent or option
the localized orbitals are printed. Note: Boys localization
can only be performed using the LOCALI command. The program will use
the Boys orbitals if they are found in the orbital record and the PIPEK option
is absent or option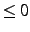 .
.
- SAVORB=record
- Allows the localized and projected
orbitals to be saved in record=name.ifil for later use
(e.g. plotting). The two orbital
sets are stored in the same dump record and can be restored at later stages
using
ORBITAL,record,[TYPE=]LOCAL or
ORBITAL,record,[TYPE=]PROJECTED, respectively.
- DOMONLY=value
- If value only domains are made, but no
energy is computed. This can be used to check and save the domains for later use.
- DSTMLT=level
- Determines the expansion level of the multipole expansion of distant pairs
(e.g. 1 means dipole approximation,
2 quadrupole approximation and so on).
The default for MULTP is 3.
- INTERACT
- Automatically determine individual molecules in a calculation and
set appropriate pair lists for computing interaction energies. See section 28.9.8
for more details.
- Parameters for energy partitioning:
- IEPART=value
- enables/disables energy partitioning.
iepart=0: Energy partitioning is disabled.
iepart=1: Energy partitioning is enabled.
iepart=2: Energy partitioning is enabled. Additionally, a list of all pair energies
and their components is printed.
- EPART=cutoff
- Cutoff parameter to determine individual monomers in a
cluster (i.e. centre groups). Should be somewhat larger than the
largest intramolecular bond length (given in a.u.).
- Miscellaneous options:
- SKIPDIST=skipdist
- Test-parameter. Its value should only affect the efficiency
but not influence the results.
skipdist=-1: Weak and distant pairs are set to zero after MP2 but are not eliminated
from the pair list and not skipped in any loop.
skipdist=0: No pairs are deleted from pair list, but weak and distant pairs are skipped
in the loops were appropriate.
skipdist=1: Very distant pairs are neglected from the beginning.
Distant pairs are eliminated after MP2.
skipdist=2: As skipdist=1, but also weak pairs are eliminated after MP2.
skipdist=3: As skipdist=2, but distant pairs are eliminated from the operator list in case
of LMP2 with multipole approximations for distant pairs. This is the default.
- ASYDOM=jiterm
- Experimental test parameter.
Enables the use of asymmetric domains for distant pairs.
The asymmetric domain approximation supplements the multipole approximation
for distant pairs, as it suppresses the treatment of configurations
for which no integrals can be computed by multipole expansion. This
leads to computational savings and improved numerical stability.
jiterm=0: Disable asymmetric domains.
jiterm=-1: Enable asymmetric domains (default).
jiterm=-2: Enable a variation of the asymmetric domain formalism:
Exchange operators will initially be projected to the asymmetric domain
instead of simply packed.
- LOCSING=locsing
- If locsing.ne.0, the single excitations use the full space, i.e.,
they are not treated locally. This is only works for LOCAL=1.
- MAXANG=lmax
- The purpose of this experimental option is to reduce
the basis set sensitivity of the Boughton-Pulay (BP) method for domain selection.
Only basis functions with angular momentum up to lmax-1 are included when computing
the overlap of the approximate and exact orbitals. For example, MAXANG=2 means to
omit all contributions of
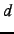 ,
, 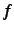 and higher angular momentum functions. To obtain reasonable
domains, the value of THRBP must often be reduced (to 0.97 or so). This
option should only be used with care!
and higher angular momentum functions. To obtain reasonable
domains, the value of THRBP must often be reduced (to 0.97 or so). This
option should only be used with care!
- PIPEKAO=option
- If option
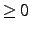 , the orbitals are localized my maximizing
the coefficients of basis functions of a given type at a given atom. Normally, this is
only useful to uniquely define degenerate orbitals in atoms. For instance, when
this option is used to localize the orbitals for a dimer like (Ar)
, the orbitals are localized my maximizing
the coefficients of basis functions of a given type at a given atom. Normally, this is
only useful to uniquely define degenerate orbitals in atoms. For instance, when
this option is used to localize the orbitals for a dimer like (Ar)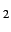 at a very
long distance, clean
at a very
long distance, clean 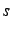 ,
, 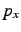 ,
, 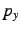 , and
, and 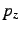 atomic orbitals will be obtained.
It is not recommended to use this option for molecular calculations!
atomic orbitals will be obtained.
It is not recommended to use this option for molecular calculations!
- NONORM=value
- Determines if projected functions are normalized (not recommended).
value=-1: projected orbitals are normalized before redundancy check.
value=0: projected orbitals are normalized after redundancy check (default).
value=1: projected orbitals are normalized in redundancy check, afterwards unnormalized.
value=2: projected orbitals are never normalized (default in gradient calculations).
- LMP2ALGO=value
- If nonzero, use low-order scaling method in LMP2 iterations.
Values can be 1, 2, or 3, and 3 is usually fastest if large basis sets are used.
- OLDDEF=value
- For compatibility with older versions: if nonzero, revert to
old defaults. Options set before this may be overwritten.
- Thresholds:
- THRPIP=thresh
- Threshold for Pipek-Mezey localization. The localization is
assumed to be converged if all
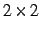 rotation angles are smaller then thresh. The
default is
rotation angles are smaller then thresh. The
default is 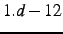 . It can also be modified globally using GTHRESH, LOCALI=thresh.
. It can also be modified globally using GTHRESH, LOCALI=thresh.
- THRORB=thresh
- Threshold for eliminating functions from pair domains whose norm
is smaller then thresh after projecting out the occupied space. The default is throrb=1.d-6.
- THRLOC=thresh
- Threshold for eliminating redundant basis functions from pair
domains. For each eigenvalue of
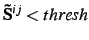 one function is deleted. The default
is 1.d-6. The method
used for deleting functions depends on the parameters IDLEIG and IBASO.
one function is deleted. The default
is 1.d-6. The method
used for deleting functions depends on the parameters IDLEIG and IBASO.
- THRMP2=thresh
- Threshold for neglecting small fock matrix couplings in
the LMP2 iterations (default 1.d-8). Specifying a larger threshold speeds up the
iterations but may lead to small errors in the energy. In the initial iterations, a larger
threshold is chosen automatically. It is gradually reduced to the specified final value during the iterations.
- THRCOR=thresh
- Threshold for deleting projected core orbitals. The functions are
only deleted if their norm is smaller than thresh (default 0.1)
The thresholds can also be specified on the THRESH directive.
Next: 32.6 Options for selection
Up: 32 LOCAL CORRELATION TREATMENTS
Previous: 32.4 Summary of directives
molpro@molpro.net
Oct 10, 2007