Next: 16.3.2 XYZ input
Up: 16.3 Geometry specifications
Previous: 16.3 Geometry specifications
16.3.1 Z-matrix input
The general form of an atom specification line is
[group[,]]atom, 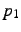 ,
, 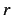 ,
, 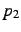 ,
, 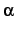 ,
, 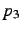 ,
, 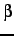 ,
, 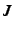
or, alternatively,
[group[,]]atom, , 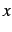 ,
, 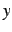 ,
, 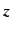
where
- group
- atomic group number (optional). Can be used if different basis sets are used for
different atoms of the same kind. The basis set is then referred to by this group number and not by
the atomic symbol.
- atom
- chemical symbol of the new atom placed at position
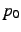 . This may optionally be appended (without blank)
by an integer, which can act as sequence number, e.g., C1, H2, etc. Dummy
centres with no charge and basis functions are denoted either Q or X, optionally appended
by a number, e.g, Q1; note that the first atom in the z-matrix must not be called X,
since this may be confused with a symmetry specification (use Q instead).
. This may optionally be appended (without blank)
by an integer, which can act as sequence number, e.g., C1, H2, etc. Dummy
centres with no charge and basis functions are denoted either Q or X, optionally appended
by a number, e.g, Q1; note that the first atom in the z-matrix must not be called X,
since this may be confused with a symmetry specification (use Q instead).
- atom to which the present atom is connected. This may be either a number n,
where
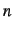 refers to the 'th line of the Z-matrix, or an alphanumeric string as specified
in the atom field of a previous card, e.g., C1, H2 etc. The latter form
works only if the atoms are numbered in a unique way.
refers to the 'th line of the Z-matrix, or an alphanumeric string as specified
in the atom field of a previous card, e.g., C1, H2 etc. The latter form
works only if the atoms are numbered in a unique way.
- Distance of new atom from . This value is given in bohr, unless ANG has been
specified directly before or after the symmetry specification.
- A second atom needed to define the angle
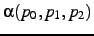 . The same rules hold for the
specification as for .
. The same rules hold for the
specification as for .
- Internuclear angle
. This angle is given in degrees and must be in the
range
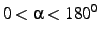 .
.
- A third atom needed to define the dihedral angle
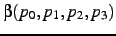 . Only applies if
. Only applies if 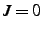 ,
see below.
,
see below.
- Dihedral angle
in degree. This angle is defined as the angle between the
planes defined by
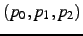 and
and 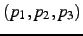 (
(
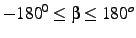 ). Only
applies if , see below.
). Only
applies if , see below.
- If this is specified and nonzero, the new position is specified by two bond angles rather than
a bond angle and a dihedral angle. If
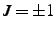 , is the angle
, is the angle
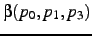 . If
. If
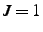 , the triple vector product
, the triple vector product
![$({\bf p}_1-{\bf p}_0) \cdot [({\bf p}_1-{\bf p}_2)
\times ({\bf p}_1-{\bf p}_3)]$](img188.gif) is positive, while this quantity is negative if
is positive, while this quantity is negative if 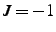 .
.
- x,y,z
- Cartesian coordinates of the new atom. This form is assumed if
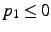 ;
if
;
if 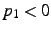 , the coordinates are frozen in geometry optimizations.
, the coordinates are frozen in geometry optimizations.
All atoms, including those related by symmetry transformations,
should be specified in the Z-matrix.
Note that for the first atom, no coordinates need be given,
for the second atom only 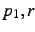 are needed, whilst for the
third atom
are needed, whilst for the
third atom 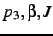 may be omitted.
The 6 missing coordinates are obtained automatically by the
program, which translates and re-orients the molecule such that
the origin is at the centre of mass, and the axes correspond to
the eigenvectors of the inertia tensor (see also CHARGE option
above).
may be omitted.
The 6 missing coordinates are obtained automatically by the
program, which translates and re-orients the molecule such that
the origin is at the centre of mass, and the axes correspond to
the eigenvectors of the inertia tensor (see also CHARGE option
above).
Once the reorientation has been done, the program then looks for
symmetry (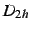 and subgroups), unless the NOSYM option
has been given. It is possible to request that reduced symmetry
be used by using appropriate combinations of the
options X,Y,Z,XY,XZ,YZ,XYZ. These specify symmetry operations,
the symbol defining which coordinate axes change sign under the
operation. The point group is constructed by taking all combinations
of specified elements.
If symmetry is explicitly specified in this way, the program
checks to see that the group requested can be used, swapping the
coordinate axes if necessary.
This provides a mechanism for ensuring that the same point group
is used, for example, at all points
in the complete generation of a potential
energy surface, allowing the safe re-utilization of neighbouring
geometry molecular orbitals as starting guesses, etc..
and subgroups), unless the NOSYM option
has been given. It is possible to request that reduced symmetry
be used by using appropriate combinations of the
options X,Y,Z,XY,XZ,YZ,XYZ. These specify symmetry operations,
the symbol defining which coordinate axes change sign under the
operation. The point group is constructed by taking all combinations
of specified elements.
If symmetry is explicitly specified in this way, the program
checks to see that the group requested can be used, swapping the
coordinate axes if necessary.
This provides a mechanism for ensuring that the same point group
is used, for example, at all points
in the complete generation of a potential
energy surface, allowing the safe re-utilization of neighbouring
geometry molecular orbitals as starting guesses, etc..
Next: 16.3.2 XYZ input
Up: 16.3 Geometry specifications
Previous: 16.3 Geometry specifications
molpro@molpro.net
Oct 10, 2007